
Small molecules form the basis for the majority of commercial drugs. These chemical entities work by binding to proteins in the target organism and modulating their function. Computer aided drug discovery attempts to facilitate the development of these molecules by computationally predicting their structure and activity, potentially allowing the development of new drugs which couldn’t be produced by other means. Docking, in particular, is an optimization process to predict the structure of the stable interaction complex between target proteins and small molecules. This task is complicated by the high flexibility of both the receptor and the ligand.
The Meiler lab is the main developer of RosettaLigand [1], [2] , a docking program which explicitly considers such flexibility. By constantly improving our docking algorithms [3], we gain further insight in the activation or inhibition of relevant drug targets.
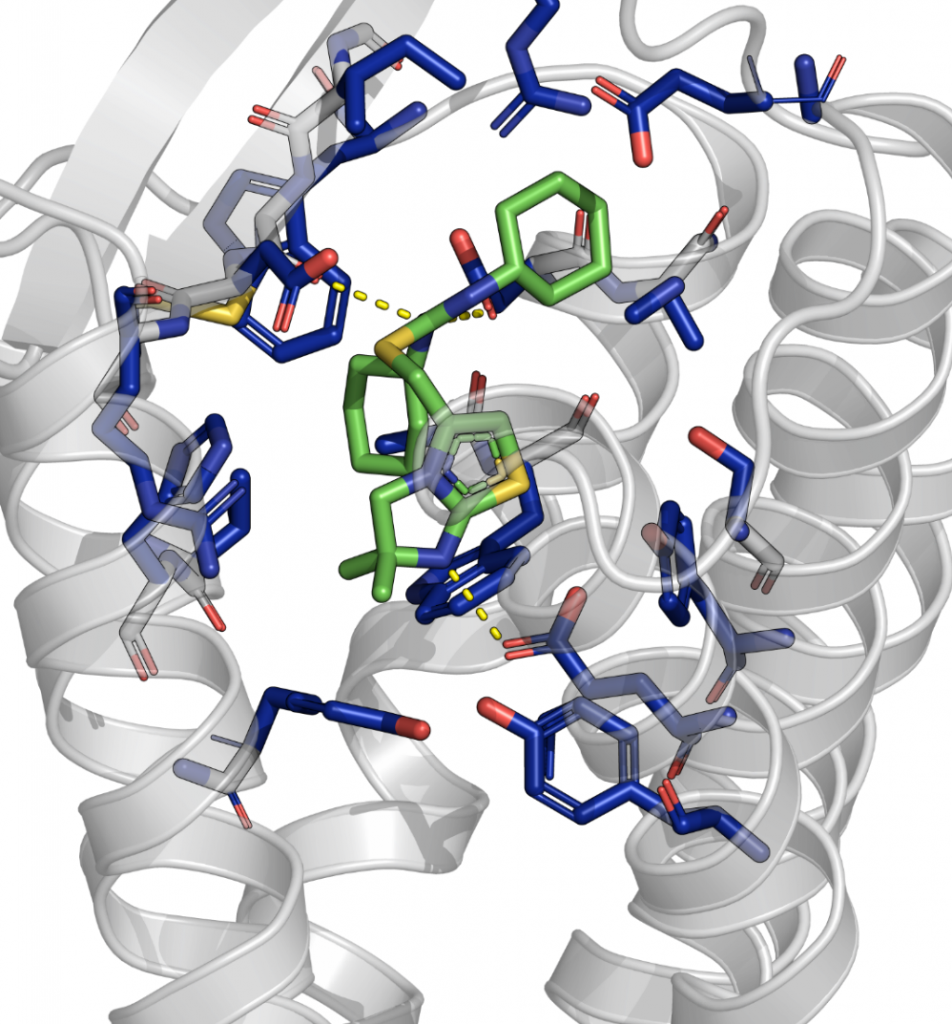
Example of a docked small molecule binder in a G protein-coupled receptor. The ligand is colored in green with hetero-atoms in blue, red and yellow, the interacting residues of the protein are highlighted, and hydrogen bonds visualized.
REFERENCES:
[1] Meiler J, Baker D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins. 2006 Nov 15;65(3):538-48.
[2] DeLuca S, Khar K, Meiler J. Fully Flexible Docking of Medium Sized Ligand Libraries with RosettaLigand. PLoS One. 2015 Jul 24;10(7):e0132508.
[3] Smith ST, Meiler J. Assessing multiple score functions in Rosetta for drug discovery. PLoS One. 2020 Oct 12;15(10):e0240450.