
De novo protein folding is a very difficult problem in structural biology. This technique works to predict protein tertiary structure from its amino acid sequence. This is done in cases where there is no template structure available (thus, we cannot do homology modeling). Because of how challenging this problem is, it is suitable for small, simple proteins.
A video tutorial can be found here: https://www.youtube.com/watch?v=wIDk5BQtM3w
A demo from Rosetta Commons can be found here: https://www.rosettacommons.org/demos/latest/tutorials/denovo_structure_prediction/Denovo_structure_prediction
The method outlined shows you how to predict protein structures with an amino acid sequence using the Rosetta AbinitioRelax protocol.
Part I – Prepare your input files
Part II – Run the Rosetta AbInitioRelax application
Part III – Analyze your results
Part IV – Adding Experimental Data
- Input Files
table {
border-collapse: collapse;
width: 90%;
}
tr {
border-bottom: 1px solid #ddd;
}
tr.last{
border-bottom: none;
}
img {
display: block;
margin-left: auto;
margin-right: auto;
}
Necessary: Optional: - Protein amino acid sequence (FASTA)
- Secondary structure prediction
- Fragment library files
- Rosetta Options file
- Native Structure (PDB)
- Constraints file – from experimental data
- Save your protein sequence to a file in FASTA format.
- Prepare fragment files by generating on your local machine OR submitting your FASTA file to the Robetta server: <a href="http://robetta.bakerlab.org/ " target="_blank">http://robetta.bakerlab.org/
- If available, copy the reference structure (usually native structure) into your input file directory to compare generated models to known native structure.
- The options file for Rosetta Abinitio contains all the flags needed to run your application. An example file is provided below (taken from the Rosetta workshop tutorial by the Meiler Lab).

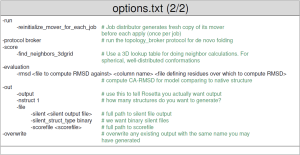
- Run:
$> ../../../main/source/bin/AbinitioRelax.default.linuxcgccrelease @input_files/options - This will take 10-20 minutes per structure. For an actual production run, 10,000 to 100,000 models need to be generated.
- Plot score vs rmsd
- Extract best scoring model
- Cluster analysis to sort structure by similarity.
Experimental data can improve prediction outcomes.
Below is a sample constraints file for EPR data (RosettaEPR) or NMR data (RosettaNMR) [taken from Rosetta workshop tutorial by Meiler Lab].
